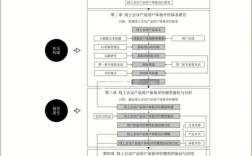
我们可以将国际药学研究中的杂质相关因素系统地归纳为以下几个核心方面,我将其称为 “杂质研究的五大核心因子”。

(图片来源网络,侵删)
国际药学研究杂质 五大核心因子
这五大因子相互关联,共同构成了一个完整的杂质控制和管理体系。
杂质的分类与来源
这是杂质研究的基础,首先要明确杂质是什么,从何而来,根据国际人用药品注册技术协调会 的指导原则,杂质主要分为以下三类:
-
有机杂质
- 来源:
- 起始物料、中间体或副产物:在合成过程中引入。
- 副反应产物:合成条件(如温度、pH、催化剂)改变时产生。
- 降解产物:药物在光照、氧化、水解、热等条件下分解产生。
- 试剂、配体或催化剂残留:合成中使用的化学物质。
- 特点:化学结构多样,是杂质研究的重点和难点。
- 来源:
-
无机杂质
 (图片来源网络,侵删)
(图片来源网络,侵删)- 来源:
- 试剂、配体、催化剂:金属(如Pd, Pt, Ni, Cu)。
- 无机盐:反应中产生的(如氯化物、硫酸盐)。
- 滤助剂或活性炭。
- 设备带来的杂质(如铁锈)。
- 特点:通常已知其化学性质,可通过药典中的通用方法(如炽灼残渣、重金属检查)进行控制。
- 来源:
-
残留溶剂
- 来源:在原料药或制剂的生产过程中使用的有机挥发性化学物质。
- 分类:根据ICH指导原则分为三类:
- 第一类(溶剂1):已知为人体致癌物、强烈怀疑对人有致癌物、环境危害物(如苯、四氯化碳),必须严格限制,通常要求 ppm 级。
- 第二类(溶剂2):非遗传毒性致癌物、其他有严重毒性的溶剂(如氯仿、甲苯),限制使用。
- 第三类(溶剂3):低毒溶剂(如乙醇、丙酮),通常有较高的限度。
- 控制:通常使用气相色谱法进行定性和定量。
杂质的鉴定与表征
在确定了杂质来源后,必须鉴定出它们是什么,尤其是那些含量超过鉴定阈值的杂质(ICH Q3A/B规定)。
- 鉴定阈值:
- 原料药:≥ 0.10%
- 制剂:≥ 0.15%
- 鉴定方法:
- 色谱-质谱联用技术:如 LC-MS (液相色谱-质谱联用) 和 GC-MS (气相色谱-质谱联用),是鉴定未知杂质结构的最强大工具。
- 核磁共振波谱:用于确定杂质的精确化学结构,特别是当质谱数据不足以确定结构时。
- 高分辨质谱:提供精确的分子量,帮助确定分子式。
- 对照品:通过与已知杂质的标准品比对来确定结构。
杂质的控制策略与限度
这是杂质研究的核心目标,即制定科学、合理的质量标准来控制杂质水平。
- 控制策略:
- 工艺控制:通过优化合成工艺(如改进反应条件、纯化步骤)来减少或消除杂质。
- 中间体控制:对关键中间体进行质量监控,防止杂质在后序步骤中累积。
- 原料药/制剂质量标准:在放行标准中设定杂质的限度。
- 限度设定依据:
- 报告阈值:≥ 0.05% (原料药) 或 ≥ 0.10% (制剂),需在申报资料中报告。
- 鉴定阈值:如上所述,超过此限度的杂质必须进行鉴定。
- 质控阈值:通常设定为鉴定阈值的一半,是日常生产和放行控制的标准。
- 分析方法:
- 高效液相色谱法:用于分离和测定大多数有机杂质。
- 气相色谱法:用于测定残留溶剂。
- 薄层色谱法:用于一些简单的杂质检查。
- 离子色谱法:用于测定无机阴离子杂质。
- 分析方法学验证:所有用于杂质控制的分析方法都必须经过验证,确保其 专属性、准确度、精密度、线性、范围和耐用性 符合要求。
稳定性研究中的杂质行为
杂质研究贯穿药品的整个生命周期,稳定性研究是其中至关重要的一环,目的是考察药品在储存期间杂质的产生情况。

(图片来源网络,侵删)
- 研究目的:
- 确定药品的储存条件和有效期。
- 识别由光照、温度、湿度等条件引起的降解途径和降解产物。
- 证明所选的包装材料能有效保护药品,防止杂质过度增加。
- 研究要求:
- 根据ICH Q1A(R2)指导原则,对原料药和制剂进行加速试验和长期试验。
- 在每个时间点取样,使用经过验证的HPLC等方法进行杂质谱分析。
- 关注任何新出现的杂质或已有杂质含量的显著增加,并与质量标准进行比较。
法规指导原则与全球合规性
国际药学研究必须遵循全球公认的法规框架,以确保药品在不同国家都能被批准上市。
- 核心指导原则:
- ICH Q3A(R2):原料药中的杂质。
- ICH Q3B(R2):制剂中的杂质。
- ICH Q3C(R8):残留溶剂。
- ICH Q1A(R2):新原料药和新制剂的稳定性试验。
- ICH Q6A:质量化学部分。
- 区域法规:
- 美国:遵循美国药典 和美国食品药品监督管理局 的cGMP及指导原则。
- 欧盟:遵循欧洲药典 和欧洲药品管理局 的要求。
- 中国:遵循中国药典 和国家药品监督管理局 的法规。
- 合规性要求:
- 在新药申报(如IND, NDA, MAA)时,必须提供全面的杂质研究资料,包括杂质谱分析、鉴定报告、分析方法学验证数据和稳定性研究结果。
- 任何生产工艺的重大变更,都需要评估其对杂质水平的影响,并可能需要补充研究。
国际药学研究中的杂质因子是一个系统性的科学和管理体系。
| 核心因子 | 关键问题 | 主要工具/方法 |
|---|---|---|
| 分类与来源 | 杂质是什么?从哪里来? | 工艺分析、文献调研 |
| 鉴定与表征 | 杂质的结构是什么? | LC-MS, NMR, HRMS |
| 控制策略与限度 | 如何控制?限度是多少? | HPLC, GC, 方法学验证 |
| 稳定性研究 | 药品在储存中会产生什么新杂质? | 加速/长期试验, 杂质谱分析 |
| 法规合规性 | 如何满足全球监管要求? | ICH指导原则, USP/EP/ChP, 申报资料 |
对这五大因子的深刻理解和严格执行,是确保药物安全有效、获得全球市场准入的根本保障。